Figure 1: CREB Pathway
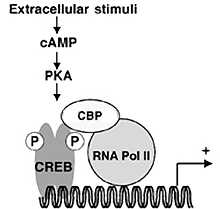
Figure 2: Equilibrium States of KIX
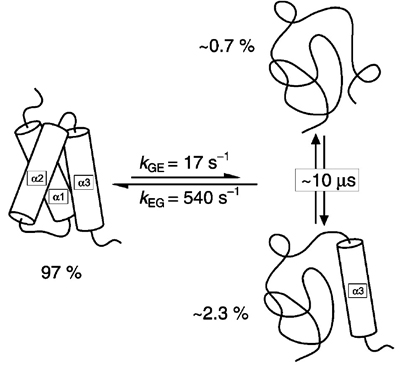
Figure 3: pKID Docking to KIX
You are using a web browser that is not fully supported by this website. Some features may not work as intended. For the best experience, please use one of the recommended browsers.
Opening Image: Average NMR structure of the KIX domain of CREB-binding protein.
This tutorial explores the binding of two intrinsically disordered proteins, pKID and c-Myb, to KIX, one of many protein-binding domains of the transcription factor CBP. The NMR structures are riveting and illustrate a number of key concepts in structural biochemistry, including the role of cooperative interactions in protein folding and molecular recognition, the importance of protein phosphorylation in regulation, and the concept of competition when two ligands bind to the same site.
Protein kinases catalyse phospho-transfer reactions from ATP to serine, threonine or tyrosine residues in target proteins and provide key mechanisms for control of numerous cellular signaling pathways and metabolic functions. The eukaryotic protein kinases represent one of the largest protein super-families; the human genome encodes about 1000 protein kinases.
Protein phosphorylation is important for several reasons. First, the phosphate group introduces two negative charges at a single location on the protein, in contrast to a charge of -1 for glutamate or aspartate. Consequently, phosphorylation can result in changes in the organization of local regions of protein structure through electrostatic interactions which in turn can result in long-range allosteric changes. Furthermore, phosphorylation is reversible through the action of protein phosphatases that catalyze the hydrolysis of the phosphate linkage to the protein. Since both protein kinases and phosphatases can themselves be regulated either by phosphorylation and dephosphorylation or by allosteric ligands, the degree of regulation by this mechanism is incredible. Finally, signal transduction through protein kinase pathways may also require protein interaction motifs which are capable of recognizing phosphorylated amino acids. That is the subject of this lesson.
One of the best understood examples of transcriptional regulation through cell surface receptor signaling is the adenylate cyclase pathway. The binding of signal molecules to receptors in the cell membrane results in an increased synthesis of cytosolic cAMP. The rise in this second messenger affects the transcription of genes which are located near a specific DNA sequence called a cAMP response element (CRE). The catalytic subunit of Protein Kinase A, released when cAMP levels rise, enters the nucleus and phosphorylates a nuclear protein, the CRE-binding protein (CREB), which binds CRE specifically. See .
CREB is a member of the leucine zipper family of transcription factors and binds CRE as a dimer through its DNA-binding domains. cAMP does not work by altering the DNA binding of CREB; rather phosphorylation of CREB alters the conformation of a region called the kinase-inducible domain (KID). This modification allows the phosphorylated domain (pKID) to interact with the KIX domain of a coactivator called CREB-binding protein (CBP). In turn, formation of the CRE-CREB-CBP complex switches certain genes on or off. As detailed below, NMR studies of the small KIX and KID fragments and the pKID-KIX complex have shown why phosphorylation of CREB is required for binding of CRB.
KIX is a small fragment of CRB; it is a 3-helix bundle. Toggle between and models of the NMR structure of KIX.
the NMR ensemble, i.e., representive configurations of KIX in solution. Each model in the ensemble corresponds to a structure calculated from a different, randomly generated starting structure. The lines in various regions of the model coalesce if their motion is limited. The greater the range of thermal motion, the farther the lines fan out. Note the following RMSD deviations from the average structure, then forget the actual numbers:
Numbers are boring, so let's look at an animation showing 16 of the best representative structures by clicking . You can use the buttons at the top of the page to toggle between models during the animation.
The frayed ends of the α-helices are typical features in NMR structures of proteins. The folding of a random polypeptide chain into the α-helix compacts the chain, thereby expelling water while forming intrasegment peptide hydrogen bonds and exquisite van der Waals contacts. These factors promote helix formation but are opposed by the entropic cost of freezing the main chain into a single conformation. Consequently, most α-helices found in proteins are not helical in the absence of interactions with other segments of the protein. The side chains provide favorable interactions but at the cost of side chain conformational entropy. However, side chains are never completely frozen in a protein, especially those at the surface of the folded protein. Let's repeat the animation, only this time with spacefilled models. Click to start.
When KIX encounters a specific binding partner, the formation of a stable complex occurs through ordering of the side chains at the interface to create complementary surfaces. In the next section we'll examine the exquisite complementarity in the pKID-KIX complex.
The NMR structure was determined at 27°C. But even at that temperature, KIX is in equilibrium with two unfolded states. See .
The borderline between order and disorder in the 3-helix bundle is indeed narrow; the isolated KIX fragment is completely disordered at 37°C.
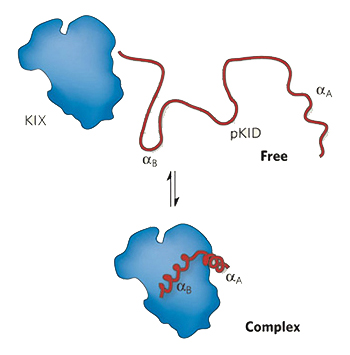
Experiments in which Ser-133 of the kinase activated domain (KID) of CREB protein is converted to Ala-133 show that the Ser residue is critical to the positive regulation of the transcriptional activity of CREB protein by Protein Kinase A. The KID fragment containing residues 119-146 is completely disordered in the absence of KIX. Phosphorylation of Ser-133 to give pKID results in the formation of a stable pKID–KIX complex in which pKID has folded into two α-helices that are at right angles to each other. The NMR structure of the complex provides an in-depth understanding of the noncovalent interactions stabilizing the complex. See .
The pKID-KIX Complex. pKID and KIX are shown in lime and cyan, respectively. The phosphoserine-133 residue is shown in CPK colors. NMR structure determined by Peter Wright's group.
The molecular of KIX is color coded to show the electrostatic potential (negative, neutral, positive). Surprisingly, the phosphate group of pKID is not juxtaposed against a postively charged region.
The docking site for pKID is a shallow groove in the surface of KIX. the side chains that make contact with pKID. Nine hydrophobic side chains (gold) and two charged side chains (magenta) of KIX make contacts with pKID.
It is important to remember that the phosphorylated kinase-inducible domain is disordered in the absence of the KIX domain. Thus binding to KIX stabilizes the two α-helices of pKID. Study the complex carefully and note the following points:
There are four strong hydrogen bonds to the exposed phosphate group of pSer-133. Phosphoserine is labeled [SEP] in JSmol.
First, note the intramolecular hydrogen bond between one of the phosphate oxygens and the hydrogen atom of the N–H amide group of pSer-133 (the α-carbon of pSer-133 is shown in dark gray).
Return to the spacefilling model. KIX and pKID are shown in cyan and lime, respectively.
Arg-131 forms a hydrogen-bonded ion pair with the phosphate group. These two intramolecular hydrogen bonds make an appreciable contribution to the stability of the ion pair.
There is only one intermolecular hydrogen bond between the phosphate group and KIX.
Tyr-658 is the only group of KIX to interact directly with the phosphate group.
Two residues of KIX, Lys-606 and Glu-655, form ion pairs with three charged groups of pKID. [Note: Residues in the 600's comprise the KIX domain, whereas residues in the pKID chain are in the 100's.]
Lys-606 forms charged hydrogen bonds with Asp-140 and Asp-144, i.e., hydrogen-bonded ion pairs.
Glu-655 forms an ion pair with Arg-124 (no hydrogen-bond).
the hydrogen bonds between the ion pairs as dashed green lines.
These ion pairs can have a subtle but significant effect on the stability of the pKID-KIX complex. However, the thermodynamics of charge-charge interactions has proven to be complex: the cost of striping off the hydration shells may be as large or larger than the electrostatic attraction between the opposite charges. Nevertheless, the steering effect of these ion pairs is important in stabilizing the complex.
The binding of pKID to KIX also involves critical nonpolar interactions at the docking site. KIX positions nine nonpolar groups at the pKID-KIX interface.
the five side chains of pKID that make nonpolar contacts with the nonpolar groups of KIX. ball-and-stick model for a better look at the interactions.
The α-helical regions of pKID in the complex with KIX extend from residue 120 to 129 and residue 134 to 144. The two helices are at right angles to each other and both make significant contacts with KIX. The hydrophobic residues Leu-128, Ile-137, Leu-138, and Leu-141 in CREB have been shown to be essential for CBP binding. Alanine mutagenesis indicated that these residues all contribute to CBP binding. Locate these residues in the model.
The stability of the pKID-KIX complex depends on the phosphorylated Ser residue and several adjacent hydrophobic residues. However, we can determine the energetic contribution of phosphate group by making small structural changes at position 133 in the kinase-inducible domain and measuring the dissociation constants of the complexes with KIX. The differences in binding energies, ΔΔGbind, will reflect only the contribution of the group at position 133, since all the hydrophobic interactions remain unchanged.
| KID Derivative | ΔΔGbind (kcal/mol) |
|---|---|
| Cβ–OH | 0 |
| Cβ–PO42- | -3.45 |
| Cβ–PO3(OH)- | -1.47 |
| Glu-133 | -1.62 |
In the S133E KID mutant, the contribution of Glu-133 to ΔGbind is 1.6 kcal/mol relative to the unphosphorylated Ser-133. This is clearly due to the electrostatic interaction between the guanidino and carboxylate groups. A similar contribution is observed if the charge on the phosphate in pKID is reduced to -1 by lowering the pH below 7. Placing two negative charges on the phosphate increases the binding energy by 3.4 kcal/mol relative to the unphosphorylated KID. This is surprising, since we saw above that the only direct interaction of the phosphate with KIX is a single hydrogen bond to Tyr-658. However, it is important to remember that dissociation (or association) constants depend on both kon and koff. Therefore, nature can increase the affinity of pKID for KIX in two ways, either by increasing the rate at which the complex forms or by decreasing the rate at which the complex dissociates -- or both.
The Arg-131/pSer-133 interaction probably does both. Recent molecular dynamics stimulations show that the Arg-131 interacts with pSer-133 significantly in the absence of KIX, but not with Ser-133 or even Glu-133 in the S133E mutant. Even fleeting interactions of Arg-131 with pSer-133 keep the polypeptide chain is closer to the final geometry of the complex, which certainly increases kon. Furthermore, by holding the two helices at right angles the Arg-131/pSer-133 interaction can significantly decrease koff.
c-Myb·KIX complex. The KIX domain is shown as a surface model. Only a fragment of c-Myb is shown as a trace model. spacefill.
Over a dozen proteins are known to modulate the activity of CRB through their binding to the KIX domain. We will see that the binding sites for two of these proteins, c-Myb and pKID, overlap physically. Therefore, while pKID occupies the docking site it prevents binding of c-Myb to KIX. This competition between the two ligands fine-tunes the regulation of KIX.
Two Arg·Glu ion pairs comprise the ionic interactions in the complex. spacefill.
the leucines of c-Myb in spacefill. Once again we see that leucines are very important. α-helices have a high propensity for leucine but not valine. The reason for this is that the presence of two methyl groups on the β-carbon of valine clash with the helix backbone. Leucine on the other hand has two hydrogen atoms on the β-carbon, which allows free rotation of the isopropyl group with respect to the helix backbone. Therefore, leucine is always better than valine when it comes to fitting an isopropyl group into a hydrophobic region.
Because c-Myb and pKID bind reversibly to KIX, the competition can be biased against pKID simply by increasing the concentration of c-Myb. When the concentration of c-Myb exceeds the concentration of pKID, the probability that pKID will bind to KIX is minimized and transcription via the adenylate cyclase pathway is slowed.
JSmol allows us to examine the pKID-KIX and Myb-KIX complexes in two ways:
c-Myb and pKID. Note that the backbone conformation of KIX is the same regardless of which ligand is bound.